Computational Spectroscopy
The group is involved in several collaborations with national and international research groups on the topics illustrated below.
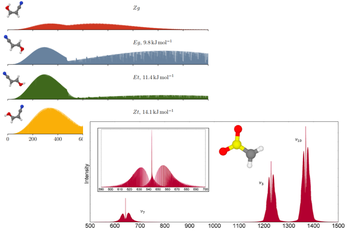
Computational studies for rotational and vibrational spectroscopy
To guide experiments in the field of rotational, ro-vibrotational and vibrational spectroscopies, it is essential to have accurate prediction of the parameters underlying these spectroscopic techniques for the molecules of interest. Indeed, the recording, the analysis and the interpretation of spectra is becoming more and more difficult as the focus is on non-stable species. Thus, computational chemistry steps-in to support and guide experimental studies, providing an irreplaceable support for the determination of spectroscopic parameters of interest, thanks to state-of-the-art methodologies.
In an astrochemistry, the spectroscopic parameters derived from the computational study are used to guide experimental observation and aid assignment. In turn, the experimental spectra are then used to perform the astronomical observation.
Some key publications can be found at the following links:
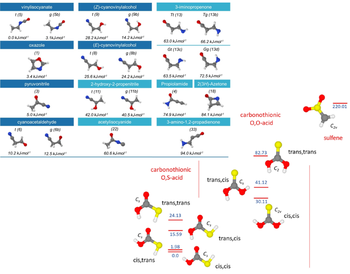
Computational Thermochemistry
Computational thermochemistry is nowadays widely used to predict and/or confirm experimentally determined thermochemical quantities. These range from conformational energies to reaction heats and dissociation energies. The role of computational thermochemistry becomes critical when unstable species, such as radiacals and ionic species are the objects of study. These are difficult to study experimentally, thus quantitative predictions are needed. For this reason, state-of-the-art methodologies are employed. These rely on the application of composite computational schemes based on coupled cluster theory. Note that accurate conformational study is the starting point for any spectroscopic study involving molecules or molecular complexes that possess large amplitude motions.
Some reference publications can be found in the links below.
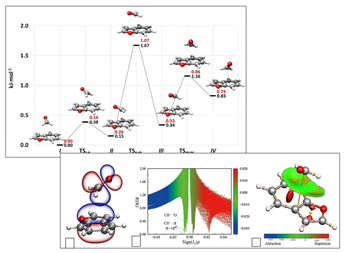
Computational studies on clusters of molecules
Covalent bonds are at the base of structural and energetic aspects of molecules, but non-bonding interactions are responsible for their unique features, such as conformational preferences, binding and recognition mechanisms, solvation processes, catalysis, and more. Non-covalent interactions have in common electrostatic aspects, which may be non-specific, such as van der Waals interactions, or may involve specific sites of molecules. This includes interactions known as hydrogen bonds, halogen bonds or lone pair-π interactions. Computational chemistry plays a very important role in understanding these weak interactions and helps in the correct interpretation of experimental results.
Examples of publications in this field are reported below.