Understanding the interplay between intramolecular and intermolecular contributions to the charge carrier propagation in organic semiconductors may guide the design of more efficient architectures.
We investigate the properties of condensed phases formed by aggregates of organic chromophores resulting in crystals or amorphous structures. These materials are at the frontier of modern applications in photonics and electronics.
Structural effects on charge transport properties are investigated in the framework of the nonadiabatic hopping mechanism. The attention is focussed on the determination of intra- and inter- molecular parameters governing the efficiency of charge transport. Charge transfer rate constants are computed within the Marcus-Levich-Jortner formalism including a single effective mode treated quantum mechanically and are injected in a kinetic Monte Carlo scheme to propagate the charge carrier in the crystal and to estimate charge mobilities at room temperature with and without the influence of an electric field.
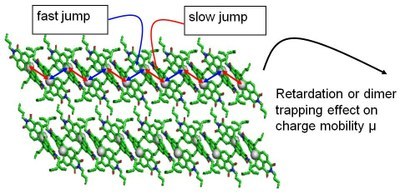
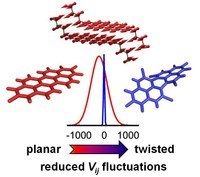
The role of low frequency crystal vibrations in modulating inter-molecular parameters is of special relevance. Thermally induced dynamical effects are investigated by means of an integrated computational approach including molecular dynamics simulations accompanied by quantum-chemical evaluation of electronic couplings, and the lattice vibrations responsible for fluctuations are identified.
One final objective is to predict the hole or electron mobility of different organic structures and their potential for the fabrication of electronic devices. In this sense the efficiency of processes at the interface between electrodes and organic materials along with the role played by intra molecular properties on these processes is another objective of this research activity.
References
